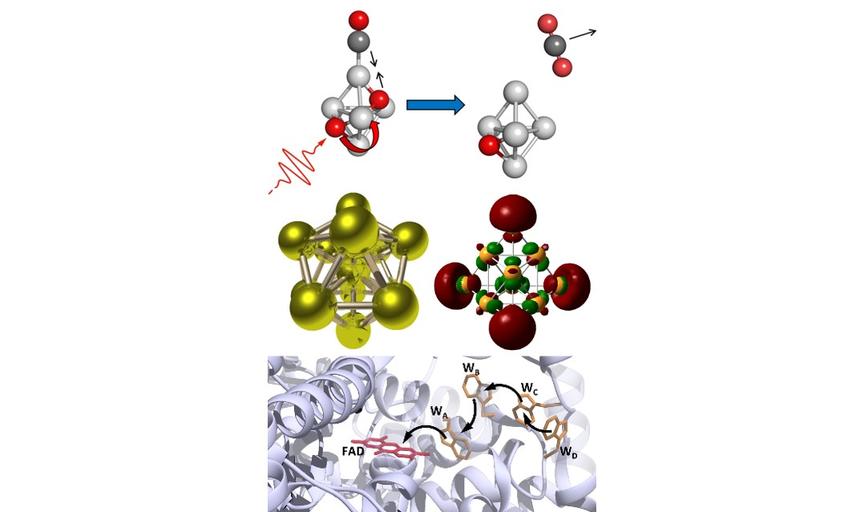
Based in Oxford University's Chemistry Department, our research interests range from the structure and reactivity of isolated (gas-phase) transition metal clusters to the photochemistry of blue-light receptor proteins relevant to animal magnetoreception.
Our work is characterised by novel instrument and technique development which enable us to make unique measurements which are interpreted with the help of quantum chemical calculations.
Please see right for a brief introduction to our group activities. Otherwise, feel free to navigate around, meet the group and find out what we do.